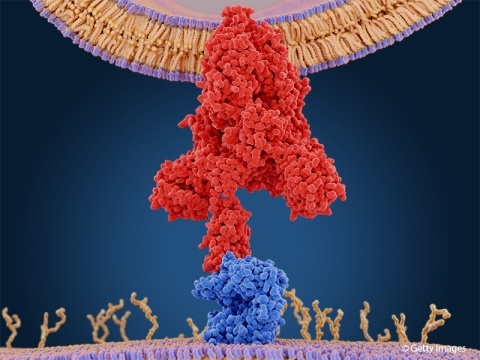
Gain insights from David Saari, Ph.D., on blending science, law, and technology in IP, highlighting the competitive edge of multidisciplinary expertise.
In this article, we speak with Mauro Mileni, Ph.D. and Chris Roth, Ph.D. about how scientists are leveraging the right technology to fill an innovative drug pipeline.
Carbon nanotubes (CNTs) hold immense promise for batteries, composites, sensors, and more. Discover emerging trends on overcoming challenges and cutting edge applications.
Data-driven technologies play a pivotal role in swift analysis and quick response to chemical threats. Optimize threat assessment and risk management strategies with quality digital solutions.
The science behind pocket-sized nuclear batteries. Discover the potential and limitations of betavoltaic batteries, from powering wearables to tackling radioactive waste.
Untangling the truth about semiconductor recycling: explore the challenges & opportunities of recycling semiconductors from designs, builds, and the future.
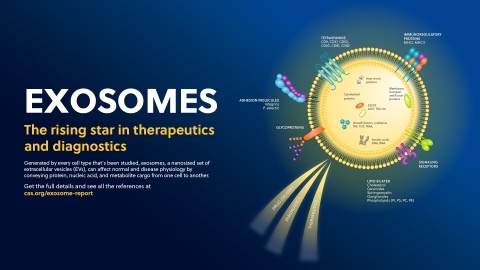
This executive summary highlights why exosomes are becoming popular in many industries. See the importance of protecting exosome IP for your next innovation.
A visual infographic shows how new innovations like biorefineries, waste repurposing, and materials are reshaping the future of sustainable fertilizers.
Stay ahead of the curve with insights from experts! Join our webinar featuring Lawrence Livermore National Lab, Oak Ridge National Lab, The Ohio State University, and CAS. Discover trends like AI’s impact in R&D, biomaterials, and tackling the undruggable.
Learn how biomarkers can improve cancer detection and treatment outcomes, and discover the latest research on pancreatic and liver cancer biomarkers. Read more.
Discover how AI in the food industry is transforming production, innovation, and sustainability. Learn how AI can help you create smarter and more efficient food solutions.
This shareable executive summary identifies emerging trends in the rapidly changing immuno-oncology field. This highlights key players, new connections, and emerging opportunities ahead.
AI re-defines natural product drug discovery. See how machine learning unlocks diverse drug candidates from plants, microbes & more, accelerating discovery
Antimicrobial-resistant infections are a global public health crisis. In a race against drug-resistant bacteria, new solutions like AI, alternative therapeutics, and innovations in materials.
In this article, we speak with Nicole Stobart, Jeff Wilson, and Mark Schmidt, about how CAS is using authority constructs to index biological molecules.
Pharmaceutical manufacturers are investigating ways to cultivate insights from dark formulations data, turning once-hidden knowledge into drug discoveries.
As the drug repurposing market grows, pharmaceutical manufacturers are investigating ways to cultivate insights from dark formulations data, turning once hidden insights into drug repurposing innovations.
Discover how Ozempic, Wegovy, and Mounjaro are part of a new class of semaglutides (GLP-1 receptor agonists) that help treat obesity and diabetes by regulating blood sugar and appetite.
Learn how photocatalysis can produce clean hydrogen from water and solar energy, and why it is crucial for decarbonization. Read the latest research on this topic.
Discover how targeted covalent inhibitors are revolutionizing cancer treatments by activating previously undruggable targets, such as RAS genes and PI3Kα. This expert webinar with Totus Medicines and CAS shows the latest trends and insights on covalent inhibitors.
Discover the emerging landscape of single walled carbon nanotubes, the new applications and approaches across industries, and what future opportunities they offer.
In this article, we speak with Iddo Friedberg, an expert in protein function prediction, about how well current prediction models are performing and more.
See how covalent inhibitors with drugs like ibrutinib and afatinib are changing cancer therapies. Identify emerging opportunities in this sharable summary.
Learn how antibody-drug conjugates (ADCs) are transforming cancer treatment with their targeted and potent action. Discover the latest trends and developments in ADC research and development.
Just in time for #Nobel season, discover the most overlooked ideas that have yet to win a Nobel Prize. From OLEDs to MOFs to mRNA therapeutics and more, these ideas have reshaped science and technology. Join the conversation on LinkedIn.
Incomplete prior art searches and analyses can lead to costly mistakes, leaving innovations and the value of intellectual property open to huge risk. Learn more here.
Explore the fascinating connection between the gut microbiome and the brain in this webinar featuring experts from CAS, Bayer, and Caltech. Learn how the gut-brain axis influences health from IBS to autism and how microbiome manipulation can offer new therapeutic strategies.
Strategies to unlock dark data in the pharmaceutical industry. Reveal data insights and accelerate innovation through digitization and knowledge management.
Discover how experts from CAS, LaJolla Institute for Immunology, and Avidity Biosciences are exploring the convergence of biology and chemistry in emerging therapeutics for cancer, vaccines, and more. The recap and slides are here.
Discover how catalysts and catalysis have transformed our world, from ancient times to modern sustainability. Learn about the latest research trends and innovations in this fascinating field of chemistry.
With consumers changing their diets for personal and health reasons, innovation to increase quality and variety creates a diverse and competitive market.
Learn how chemistry powers space exploration with rocket fuel and engine technologies. Discover the advantages and challenges of using LOX/LH2 as a propellant for the Artemis mission.
Learn how catalysts have been shaping human history and enabling sustainable chemistry in this executive summary. Discover current research trends and future opportunities in using non-noble metals as catalysts.
Learn how mRNA vaccines, which have revolutionized the fight against COVID-19, can also be used to treat cancer by harnessing the power of the immune system.
Biomaterials are re-shaping the landscape of drug delivery, diagnosis, and therapeutics, learn more about the top ten emerging trends in this space. Key materials covered include hydrogels, lipid nanoparticles, exosomes, bioinks, programmable biomaterials, and more.
This one-page executive summary on anti-aging strategies and treatments highlights emerging trends, new approaches, and future opportunities from stem cell therapies, prescription drugs, and lifestyle changes. This includes key take-aways of clinical trial pipeline analysis and a landscape view of emerging treatment strategies.
Discover the latest in anti-aging treatment strategies to promote health and longevity. Uncover the latest research on stem cell therapies, hyaluronic acid, senolytics, gene therapy, and a pipeline analysis of ongoing clinical trials in our latest article.
Discover how microplastics have infiltrated our entire world and the innovative new approaches that are being used to remove them in our latest infographic.
In this Challenges and Opportunities article, we talk with Jefferson Parker, an expert in data analytics for life sciences, about sequence analysis for drug discovery and the frontiers for AI in computational biology.
Supercapacitors vs. batteries: Is graphene the key to unlocking their full potential? Learn how graphene-based supercapacitor technologies are ready for real-world applications and what challenges they face.
This idea in brief provides an overview of the latest trends and emerging opportunities in mRNA research and development. It discusses the key benefits of mRNA technologies, the challenges that remain, the key players, and their areas of focus.
Strategies to unlock dark data in Chemistry R&D. Reveal data insights and accelerate innovation through knowledge management and data strategy planning.
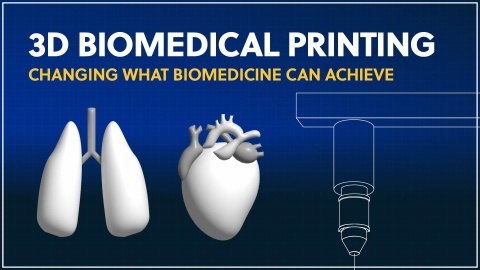
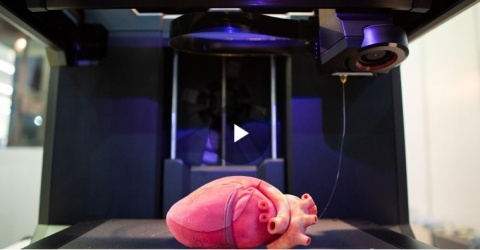
See how 3D printing is finally transforming biomedical applications in our new infographic. See how advances in materials and approaches have re-shaped the following areas: sourcing of tissues, transplants, and organs, new drug delivery methods, and advances in surgical settings and wound care.
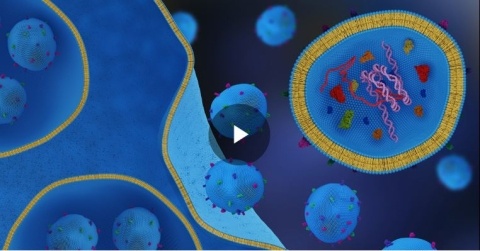
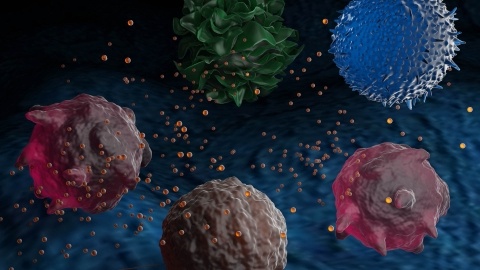
Discover how exosomes, nature's lipid nanoparticles, are reshaping drug delivery and treatment of diseases from cancer to cardiovascular disease and beyond. Learn from experts at Mayo Clinic, Direct Biologics, and Aruna Bio in our recent ACS webinar. Watch the recording and read the full Insight Report now.
Discover how RNA therapeutics are transforming medicine in our new infographic. Learn about the applications, challenges and opportunities of this cutting-edge technology in cancer, infectious diseases, liver and metabolic diseases. Find out the latest trends and data on key nanoparticles, emerging types of RNA and modified sequences in this rapidly evolving space.
This peer-reviewed journal publication in Bioconjugate Chemistry shows how PEGylated lipid nanoparticles are used to deliver mRNA vaccines and other drugs, the immunogenicity concerns with vaccines, and emerging approaches on new designs to increase efficacy and safety.
Learn how to address PEG immunogenicity, a challenge that affects the safety and efficacy of PEGylated lipid nanoparticles for drug delivery. Discover the latest R&D trends, market data, and opportunities in this cutting-edge field including COVID-19 vaccines and a wide range of current and future drugs that leverage these nanoparticles.
Learn how to overcome PEG immunogenicity, a challenge that affects the safety and efficacy of PEGylated lipid nanoparticles for drug delivery. This article reviews the current state-of-the-art in addressing PEG immunogenicity, from elucidating the factors that influence anti-PEG antibody production to developing novel drug vehicles or optimizing administration and posology.
In this edition of the CAS article series, Challenges & Opportunities in Drug Discovery, we talk with Ben Taft, the Senior Director of Chemistry at Via Nova Therapeutics, about structure-activity relationship studies and the possible frontiers for bioactivity data.
This peer-reviewed publication in the ACS Chemical Neuroscience Journal details a landscape analysis of emerging trends in the gut microbiome space. It dives deep into an overview of microbiome therapies, emerging approaches, new science, clinical pipelines, and key players in this space.
This report features a comprehensive landscape analysis of the gut microbiome, including its composition, functions, and role in health and disease. Created in collaboration with Bayer AG, this article discusses the latest research on microbiome-based therapies, microbiota, probiotics, and the wide range of health conditions it can influence, like obesity, diabetes, inflammatory bowel disease, and even mental health disorders.
The gut microbiome is a complex ecosystem of trillions of bacteria that live in our digestive tracts with health benefits like immunity, improved mental health, and therapeutic options. In recent years, there has been a growing interest in the potential of microbiome therapies to treat a variety of diseases. As our understanding of the gut microbiome continues to grow, we can expect to see more advances in microbiome therapies in the years to come.
Microplastic pollution is a global problem and can be ingested by animals and humans. They can also accumulate in soil and water, where they can disrupt ecosystems. There is a growing body of evidence that suggests that microplastics can be harmful to health. This executive summary highlights key sources of microplastic pollution, health risks, and steps that can be taken to reduce microplastic pollution
As the leading cause of drug-related deaths, fentanyl has become a public health crisis, but new breakthroughs in developing safer pain medications, reducing side effects, and developing a vaccine may reduce future deaths.
Exosomes are reshaping the future of drug delivery, therapeutics and diagnostics in cancers and beyond. This full-sized infographic details critical types of exosomes, the diseases (like cancer) being studied, and a deeper dive into the challenges that need to be addressed before its robust clinical pipeline is approved.
Understand emerging trends of 3D printing in biomedical applications and the new technologies that are reshaping personalized healthcare. From 3D printing devices to custom drug delivery and printing bioactive tissues and organs, this field is innovating rapidly with implications across the healthcare industry.
Read this peer-reviewed journal article about the synthetic organic chemistry research landscape, published in collaboration with the National Natural Science Foundation of China, the National Science Library at the Chinese Academy of Sciences, and CAS.
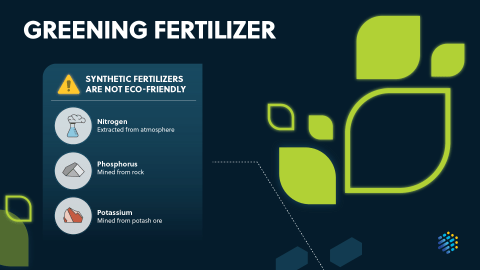
For the executive who needs to quickly understand emerging trends around sustainable agriculture, green fertilizer production, and reducing the carbon emissions of agriculture this idea in brief will provide unique insights for their R&D teams.
Discover the science behind vinyl chloride, dioxins and different remediations being developed. Explore the latest patent trends, research publications, and data challenges surrounding dioxins and vinyl chloride to understand how we can better manage hazardous material transportation in the future.
Discover how exosomes, nature's lipid nanoparticles, are reshaping drug delivery and treatment of diseases from cancer to cardiovascular disease and beyond. Learn from experts at Mayo Clinic, Direct Biologics, and Aruna Bio in our recent ACS webinar. Watch the recording and read the full Insight Report now.
For the executive who needs to quickly understand emerging trends around exosomes, lipid nanoparticles, and their impact on drug delivery, diagnostics, and therapeutics this idea in brief will provide unique insights for their R&D teams.
Learn about the latest advancements in lithium-ion battery recycling technology in this article. Discover a new approach for direct recycling that could make it possible to repair and reuse depleted batteries at a lower cost. Understand the challenges still facing the industry and the potential impact on the future of battery recycling.
Explore the limitless potential of 3D printing in biomedicine with this in-depth report. From prosthetic ears to artificial organs, learn about the latest trends and innovations. Discover the potential that 3D printing in biomedicine holds for manufacturing, forensic pathology and more.
Discover the innovative applications of biomedical 3D printing, starting with from a short history of the technology, all the way to the latest trends of 3D printing in healthcare.
Explore the possibilities of 3D printing in biomedicine with this analysis from a scientific journal. From the production of tissues and organs to personalized prostheses and medications, learn about the challenges overcome and how 3D printing in biomedicine will re-shape the future.
Stay ahead of the curve with the latest in emerging science and cutting-edge research that is accelerating the landscape of innovation. From new discoveries in synthetic biology to breakthroughs in sustainable energy, these are the breakthroughs that will reshape 2023. The experts at CAS reveal unique analysis and emerging trends from their landscape view of the world’s science.
Scientific journal review examines and analyzes the scientific and patent publication trends so the scientific community understands key breakthroughs to build upon, emerging compounds, and the challenges ahead.
Explore the path to sustainable agriculture and learn about the key role it plays in global food production. Discover how reducing, reusing, and recycling can improve fertilizer access, minimize environmental impact and dependence on finite resources, and lead to a circular bioeconomy. Learn about the latest innovations in sustainable agriculture, including smart nano-fertilizers, biorefineries, and biochar, and see how they can be used to harness the nutrient content of waste products to bolster food production and protect the environment.
Detailed landscape report on the latest trends in sustainable agriculture and fertilizer production, new opportunities and insights on the scientific advancements that will enable future innovation
Scientific journal review detailing the emerging landscape of microplastics research, publication, and patent trends. This analysis highlights new opportunities and challenges facing humanity that is awash in microplastics.
Understand the environmental threat of microplastics by exploring its impact through resources such as the 2022 WHO report on dietary and inhalation exposure to microplastics, and microplastics publications trends.
In-depth, detailed landscape report that reveals new approaches, publication trends, and ideas gaining traction in the fight against microplastics pollution.
Learn more about ACE2 the critical protein that remains the critical target of COVID-19 and so many additional viral infections. This overviews emerging trends and insights on publications, milestones, and future applications to target ACE2 proteins.
Landscape view of emerging contributors, companies, and researchers in the emerging field of exosome research. This article overviews key players in diagnostics and therapeutic fields of exosome research with a deeper look into the current clinical pipeline.
Exosomes’ unique physical properties could unlock massive changes in the fields of drug delivery and diagnostics. However, the biggest challenge for exosomes is the methodologies of isolation and purification – this article discusses the recent trends and applications of exosomes in drug delivery, diagnostics, and therapeutics.
The FDA can designate drug candidates as “breakthrough therapies” (breakthrough therapy designation or BTD) for certain drug candidates that demonstrate substantial improvement over available therapies on a clinically significant endpoint. This infographic highlights the value of this designation, key requirements, and factors that go into this designation.
Exosomes are nature’s lipid nanoparticles and their unique physical properties will unlock new applications in drug development, delivery and diagnostic applications in the future
A historical overview of the discovery of exosomes, key milestones in their rise, and why they will revolutionize the diagnostic and drug delivery fields in the future.
Ammonium nitrate has the power to feed billions, yet when stored improperly is like a bomb waiting to happen. How can we make this fertilizer safer? Learn about emerging science, safer alternatives, and best practices for safe storage and transport.
Learn more about lipid nanoparticles, their origin, role in COVID vaccines, and future applications. This overviews emerging trends and insights on publications, milestones, and future applications in vaccines, drug delivery, and non-therapeutic applications like agro-chemistry, cosmetics, and food science.
Learn more about cytokine storms and their role in both immunity and severe infection with COVID-19 and additional viral infections. This overviews emerging trends and insights on publications, milestones, and future applications to minimize the impact of cytokine adverse effects.
Learn more about the hidden greenhouse gas emissions of plants, a more sustainable approach to fertilizer production, and new approaches to make phosphorus more recyclable.
Deformulation is the process of reverse-engineering the composition of known formulations. This publication shows the value of consistent and highly structured representations in predictive models.
If you are producing fertilizers, phosphates, or any of our critical agricultural products, you know sustainability is a big challenge. Join us for the latest in phosphate recycling, sustainable ammonia production, and alternative fertilizer production. Gain insight into the latest market, scientific research, and publication trends that reveal the opportunities ahead.
Research and development teams amass huge quantities of complex data which, if exploited correctly, can be a useful information source to improve decision-making and drive innovation.
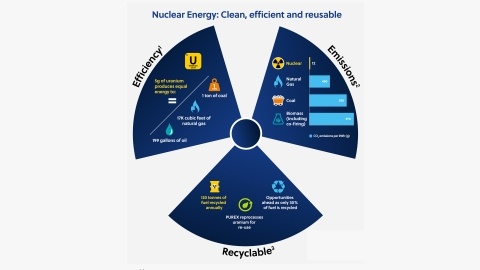
Visual infographic of nuclear energy’s key benefits of an efficient fuel source, a low emissions option (compared to coal and natural gas) and new recycling efforts that minimize nuclear waste.
Learn more about the expert panel discussion on molecular glues and targeted protein degradation. Expert panelists include Dr. Benjamin Ebert, the chair of Medical Oncology at the Dana-Farber Institute, Dr. Phil Chamberlain, the co-founder, President, and CEO of Neomorph, and Janet Sasso, an information scientist specializing in Life Sciences at CAS.
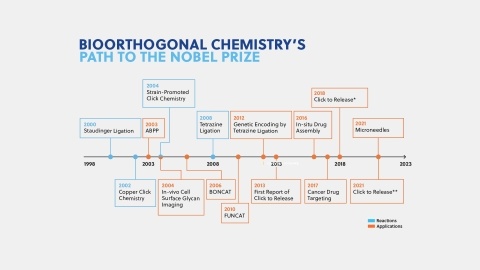
Learn about the basics of click chemistry and bioorthogonal chemistry that was recently awarded the 2022 Nobel Prize in Chemistry. Read about emerging trends, insights on publication trends, and future applications of bioorthogonal chemistry.
"When you’re finished changing, you’re finished." Business leaders likely weren't the intended audience when Benjamin Franklin uttered his now famous quote. However, his words likely resonate deeply with leaders in today's rapidly evolving marketplace who know that for their business to thrive, they must be prepared to adapt and embrace new technologies.
When mRNA (messenger RNA) vaccines are injected into a patient, tiny lipid vesicles transport mRNA molecules through the body fluids and merge with immune cells, called antigen-presenting cells (APCs). mRNA vaccines work to instruct APCs to manufacture proteins called antigens that evoke an immune response to the threat.
One approach that has gained momentum since the 1990s is green chemistry, a scientific field focused on "the invention, design, and application of chemical products and processes to reduce or eliminate the use and generation of hazardous substances.
Find out how companies improve their prediction accuracy, obtain Breakthrough Technology Status and shorten the development and review time of new drugs by leveraging CAS advanced machine learning methods.
This blog details the recently published CAS indicator of drug innovation based on the structural novelty of new molecular entities (NMEs) which may better assess innovativeness of new drugs.
Scientists at Bayer expand their understanding into new, useful chemistry by improving the predictive power of a synthesis planning model with scientist-curated reactions from CAS.
Bioorthogonal chemistry is a set of methods using the chemistry of non-native functional groups to explore and understand biology in living organisms. This review summarizes common reactions used in bioorthogonal methods, their advantages and disadvantages, and their frequency of occurrence in the literature.
Learn more about the scientific challenges, emerging technologies, and the financial and environmental implications for recycling lithium ion batteries.
Read our white paper to find out more about the advantages, limitations, and popularity of the different classes of biopolymers, and how the research and development interest in these alternatives to traditional plastic has changed over the last two decades.
This whitepaper explores challenges and opportunities for worldwide patent offices as they seek to ensure sustainability and future growth. It focuses on the application of artificial intelligence-enabled workflow solutions to enhance productivity and patent quality.
NASA’s Artemis Program has also focused attention on topics such as food safety, reliability, and nutrition in space. This blog touches on novel approaches to these problems like 3D printed space food, the use of microbes to produce nutrients, and closed-loop space farming.
It is estimated that one in five human cancers involve RAS mutations. Although once considered undruggable, this blog looks at recent advances in RAS inhibitors and emerging therapies.
The COVID pandemic revealed deficiencies in mask development and production. Innovation will be crucial in slowing the spread of new variants of the virus. There are many factors that can be improved for masks, and this article focuses on the emerging science of improving filtration and microorganism removal.
mRNA vaccines have many potential advantages over conventional vaccines in mitigating the spread and eventually ending the COVID-19 pandemic. Learn more about mRNA vaccines and how CAS is committed to supporting the fight against COVID-19.
As COVID-19 cases rise again worldwide, the Omicron subvariant BA.5 is currently the dominant strain. This blog explores some key mutations that increase transmission, evade protective antibodies, and increase the rate of (re)infection.
Targeted protein degradation is a novel and quickly expanding drug discovery strategy. It is a new way of utilizing disease-causing proteins to attack severe illnesses, which has the potential to be used among more serious illnesses, such as cancer or neurodegenerative diseases.
Artificial intelligence (AI) and machine learning have been growing exponentially over the last decade, but how has the field of Chemistry evolved with this emerging trend?
Molecular glues have been used to uncover new therapeutic agents. Because of their targeted protein degradation that holds them together, these glues have exciting potential in uncovering new drugs to fight cancer, immune diseases, and more.
Lipid nanoparticles play critical roles in mRNA vaccines for treating COVID-19, but their use is not new. Read about past, present, and future opportunities.
With recent headlines on monkeypox, learn more about the research landscape, scientific profiles, and currently available therapies. A deeper understanding of the published science enables clarity and insight on this emerging news story.
This article summarizes the results of an extensive recent analysis of carbon dioxide capture, including research trends, methods employed, and storage.
In the past decade, there has been a shift in research, clinical development, and commercial activity to utilize RNA for medicine. With the rapid success in the development of lipid–RNA nanoparticles for mRNA vaccines against COVID-19 and several approved drugs, RNA has catapulted to the forefront of drug research.
We are in the midst of a therapeutic revolution, according to the authors of a recent review article in Frontiers in Bioengineering and Biotechnology. They were commenting on the rapid expansion of RNA therapeutics in modern research and clinical development, driven in part by interest in RNA COVID-19 vaccines in the ongoing SARS-CoV-2 pandemic.
A literature search in the CAS Content Collection provides useful insights into recent and ongoing intensive research and development into green hydrogen production, storage, and utilization in fuel cells. The trends in publications suggest that hydrogen storage and fuel cells are becoming technologically mature whereas, green hydrogen production is still at an exploratory stage. Read the blog to learn more.
Many technologies have been researched with an eye toward enabling a hydrogen economy. In the field of hydron utilization in fuel cells, numerous materials have been created to target higher efficiencies and applications.
Most synthetic polymers are derived from fossil fuel resources, whose scarcity and imbalanced global distribution can affect plastics producers. Bio-based polymers, obtained from renewable biomass resources, have received wide attention in the past two decades.
While human capabilities have only increased due to recent technological and medical advances, they have significantly contributed to the release of around 830 gigatons of CO2 into the atmosphere in just the last 30 years alone.
It turns out that these intrinsically disordered proteins may be the key to better overcoming diseases in neurodegeneration, diabetes, cardiovascular disease, amyloidosis, genetic diseases, and cancer.
This blog lists and discusses the common ingredients (both in the household and scientific contexts) as well as the unique ingredients used in the Covid-19 vaccines currently in use in the United States, and also addresses the dosage levels proposed for children under five years of age.
In the ongoing pursuit of greener energy sources, lithium-ion batteries and hydrogen fuel cells are two technologies that are in the middle of research boons and growing public interest. Read this blog to learn more about the promises and challenges of these technologies.
Gut microbiota, gut flora, or microbiome are the microorganisms that live in the digestive tracts of humans and other animals. While some bacteria are associated with disease, others are particularly important for many aspects of health and mental health.
Lithium-ion batteries (LIBs) are commonly used in a variety of consumer products, including cellular phones, laptop computers, and more recently, electric and hybrid vehicles.
As the production and usage of lithium-ion batteries (LIBs) has increased exponentially, their manufacturing and disposal have become subjects of political and environmental concerns.
CAS enables views into past journal and patent publications for lithium-ion battery recycling, including processes, emerging trends, and global challenges.
Preliminary study of the Omicron variant indicates that it has evolved significantly from the original version of the virus first identified in China in 2019, which means there is an increased likelihood that it could reinfect those who have already had COVID-19 or evade the immunity generated by the current first generation of vaccines. Such a variant could drastically set back the global pandemic recovery, resulting in staggering societal and economic impacts.
With recent mRNA booster recommendations, many ask if they should get a COVID-19 booster and what the science shows? This blog explains the science of boosters, reviews current recommendations, and examines recent research.
Newly developed microfluidic fabrication technology addresses the clinical need for scalable, precise, and reproducible solutions for vaccine production.
To mark the anniversary of the industrial accident and explosion in Beirut, this CAS Insight focuses on the role of ammonium nitrate and how weak bonds in certain chemicals can lead to instability and explosions.
In recent years, AI application to chemistry has rapidly grown. While there has been numerous publicity about AI used in this manner, there hasn’t been a lot of in-depth analysis on its use and development in the chemistry field.
Lipid nanoparticles (LNPs) have emerged as promising vehicles to deliver a variety of therapeutics in the pharmaceutical industry. Liposomal drugs, based on an earlier version of LNPs, have already been approved and applied to medical practice.
This report reviews Covid-19 vaccine research, compares the underlying technologies, the use of adjuvants and delivery systems in their application, and provides a perspective on their future direction.
The Pistoia Alliance Chemical Safety Library (CSL) is an open-access database providing access to hazardous reaction information crowd-sourced from academic, industry, and government institutions around the world. Developed and hosted by CAS, the CSL supports laboratory safety.
This in-depth CAS Insights Report discusses the chemical properties of ammonium nitrate, its hazards and safety rules, and provides a useful resource for those involved in the handling and storage of this compound.
Learn how machine learning procedures and CAS COVID-19 resources, including scientific insights, open access datasets and special reports are assisting in the research to fight against COVID-19.
To identify potential anti-SARS-CoV-2 therapeutics, CAS scientists are building machine-learning predictive models to identify novel drug candidates for the viral targets 3 chymotrypsin-like protease (3CLpro) and RNA-dependent RNA polymerase (RdRp).
In support of ongoing research efforts to identify additional drugs for COVID-19, a team of CAS scientists analyzed published scientific information and created a comprehensive report focusing on proteins involved in COVID-19 and potential corresponding drug candidates. Learn how CAS has committed to leveraging their assets and capabilities to support the fight against COVID-19.
To support ongoing research and development of COVID-19 therapeutics, this report provides an overview of protein targets and corresponding potential drug candidates with bioassay and structure-activity relationship data found in the scientific and patent literature for COVID-19 or related virus infections.
In this article from The Patent Lawyer Magazine, Anne Marie Clark, Senior Search Analyst for CAS, discusses how to avoid patent pitfalls as the market for RNAi-derived therapeutics grows.
A critical step in the race to develop treatments for COVID-19 is to gain a clear understanding of how the virus enters our cells. This insight will support development of targeted anitviral treatments focused on blocking that pathway.
To provide a better understanding and comparison of the diagnostic tests available in the fight against COVID-19, CAS has produced a report summarizing the basic principles of molecular and serological assays being used in diagnostic tests for SARS-CoV-2. The report highlights recent advancements in testing technologies and provides a high-level view of currently available diagnostic tests.
To support R&D on COVID-19, CAS has produced a special report to provide an overview of published scientific information with an emphasis on patents in the CAS content collection. It highlights antiviral strategies involving small molecules and biologics targeting complex molecular interactions involved in coronavirus infection and replication.
While hosting 15-20% of Earth’s biological diversity, a substantial part of Brazil’s abundant biodiversity remains underexplored. A lack of organized information made it exceedingly difficult for researchers to search, screen or even compare relevant chemical substances. This was hindering their ability to identify new targets, build on prior discovery and drive innovation.
The Journal of Organic Chemistry article "Recent Changes in the Scaffold Diversity of Organic Chemistry As Seen in the CAS Registry" shows the pace of innovation from a structural perspective is accelerating despite the extensive reuse of a relative small number of scaffolds.
Induced-proximity target protein degradation (TPD) is a ground-breaking strategy in drug discovery that has emerged in the last decade. This CAS white paper surveys the research landscape, status of clinical trials, and discusses possible future opportunities.
Download this in-depth CAS Market Report to gain insight into the evolution of hydrogen fuel technology, the global innovation landscape, key market trends and potential applications and opportunities.