Gain new perspectives for faster progress directly to your inbox.
Les maladies neurodégénératives affectent considérablement la vie de millions de personnes à travers le monde. Ces troubles complexes sont liés à de nombreux facteurs génétiques et non génétiques, mais tous manifestent une agrégation pathologique des protéines ou des problèmes de protéostase, le processus de synthèse, de repliement, de désagrégation et de dégradation des protéines. Les maladies neurodégénératives les plus connues sont la maladie d'Alzheimer et la maladie de Parkinson, qui sont devenues plus courantes du fait de l'allongement de l'espérance de vie, qui expose de plus en plus de personnes aux effets du vieillissement. Il existe cependant un autre groupe de maladies neurodégénératives appelées maladies par expansion de polyglutamine, parmi lesquelles la maladie de Huntington, qui présentent des défis supplémentaires aux chercheurs en quête d'options de traitement.
Les maladies par expansion de polyglutamine se caractérisent par l'expansion anormale d'une répétition du trinucléotide cytosine-adénine-guanine (CAG), qui aboutit à la formation de protéines présentant un conduit de polyglutamine étendu. Ces maladies sont considérées comme rares, touchant en moyenne 1 à 10 personnes sur 100 000, mais les maladies par expansion de polyglutamine constituent le plus grand groupe de troubles neurodégénératifs monogéniques. Sans remède ni mesure préventive disponible, ces maladies sont compliquées à traiter et peuvent affecter considérablement des familles entières en raison de leur nature génétique.
Nous avons analysé CAS Collection de contenusTM, le plus grand référentiel mondial d'informations scientifiques structurées par des humains, afin de mieux comprendre les avancées actuelles de la recherche et les difficultés constantes du traitement de ces maladies graves.
Que sont les maladies par expansion de polyglutamine ?
Il existe neuf maladies par expansion de polyglutamine identifiées : la maladie de Huntington (MH) ; six ataxies spinocérebelleuses (SCA) des types un, deux, trois, six, sept et dix-sept ; l'atrophie dentato-rubro-pallido-luysienne (ADRPL) ; et l'atrophie musculaire bulbo-spinale (AMB). Toutes ces maladies sont autosomiques dominantes à l'exception de l'AMB, qui est liée au chromosome X et ne touche que les populations masculines.
Les maladies par expansion de polyglutamine présentent une corrélation inversée entre le nombre de répétitions du CAG et l'âge des patients à l'apparition de la maladie. La maladie a tendance à s'aggraver et l'âge auquel elle apparaît tend à diminuer dans les générations successives.
Les protéines impliquées dans les différentes maladies diffèrent par leur fonction et leur emplacement dans les cellules, et chaque maladie affecte différentes régions du cerveau et sous-types de neurones. Toutefois, elles présentent une caractéristique commune : la déformation radicale des neurones dans des régions spécifiques du cerveau, ce qui perturbe les fonctions vitales.
Les difficultés du traitement de la maladie de Huntington et des maladies par expansion de polyglutamine
Il n'existe actuellement aucun traitement de fond pour la maladie de Huntington ou d'autres maladies par expansion de polyglutamine, et plusieurs difficultés constantes compliquent la recherche de médicaments :
- Physiopathologie complexe : les maladies par expansion de polyglutamine impliquent des mécanismes moléculaires délicats aux multiples complexités que les chercheurs s'efforcent de mieux comprendre.
- Présentation clinique variable : ces maladies possèdent une hétérogénéité importante en ce qui concerne la présentation, l'âge de l'apparition et la progression de la maladie. Cela complique le diagnostic et le traitement.
- Absence de biomarqueurs pour le diagnostic et la surveillance : des biomarqueurs fiables sont essentiels pour le diagnostic, le pronostic et la surveillance des réponses au traitement, mais les biomarqueurs des maladies par expansion de polyglutamine sont limités.
- Multiples obstacles au développement de médicaments : les modèles animaux ne représentent pas pleinement le phénotype de la maladie humaine de ces maladies, de sorte que leur valeur est limitée dans le cas des maladies par expansion de polyglutamine. Dans le même temps, les traitements potentiels ont du mal à pénétrer la barrière hémato-encéphalique et à atteindre les neurones ciblés. Le tout complique le développement de traitements efficaces.
La recherche se développe, mais il reste beaucoup de travail
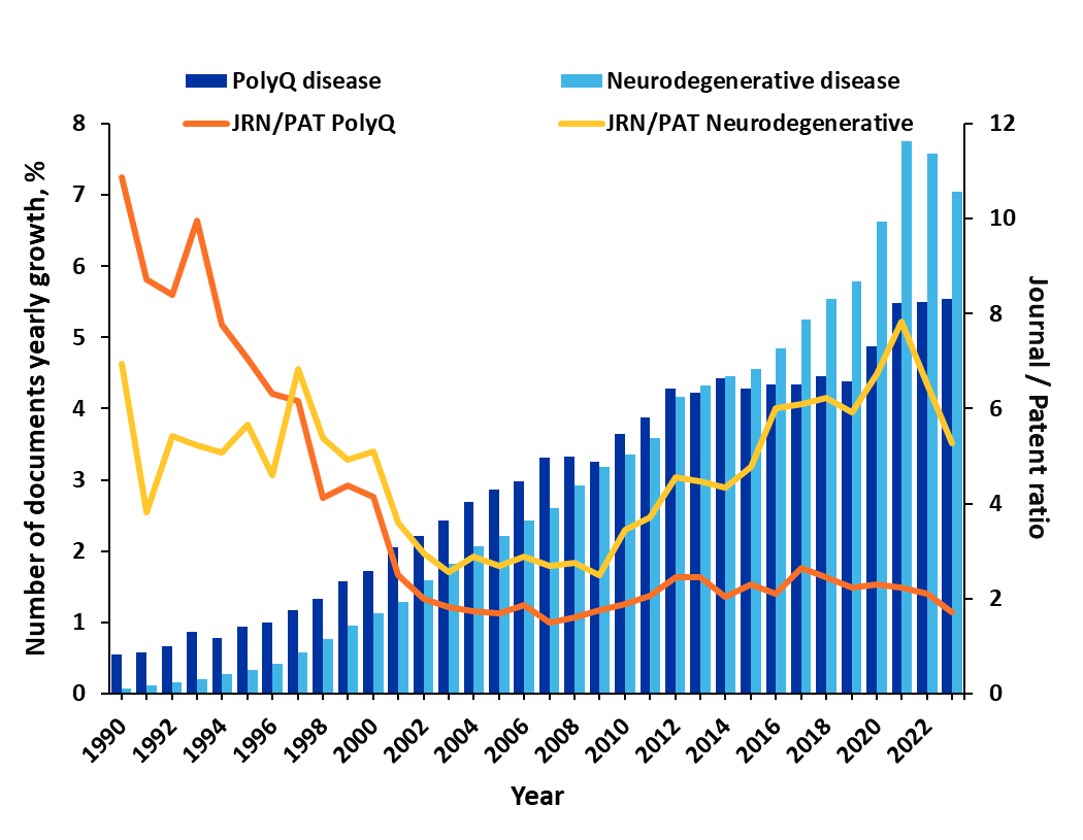
Malgré ces nombreuses difficultés, les chercheurs s'efforcent de mieux comprendre ces maladies et leurs traitements potentiels. Notre analyse de CAS Collection de contenus a révélé une augmentation de plus de 25 % des publications au sujet des maladies par expansion de polyglutamine au cours des trois dernières années seulement. Il est à noter qu'auparavant, les publications de revues dominaient la recherche avec un ratio revues-brevets de 4-5, mais que depuis 2002, les publications de brevets ont nettement augmenté. Ce ratio est désormais plus proche de 2 (voir figure 1). Cela suggère que les découvertes thérapeutiques se rapprochent de la commercialisation.
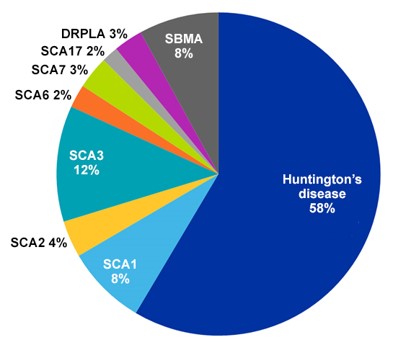
Plus de la moitié des documents liés aux maladies par expansion de polyglutamine figurant dans CAS Collection de contenus concernent la maladie de Huntington, ce qui est logique dans la mesure où il s'agit de la plus répandue des maladies par expansion de polyglutamine, avec une fréquence moyenne de trois à sept cas pour 100 000 personnes (voir figure 2). La chorée est le symptôme le plus courant de la maladie de Huntington, mais elle entraîne également des modifications cognitives et une démence. Elle est déclenchée par une expansion de la répétition du CAG de l'exon du gène de Huntington.
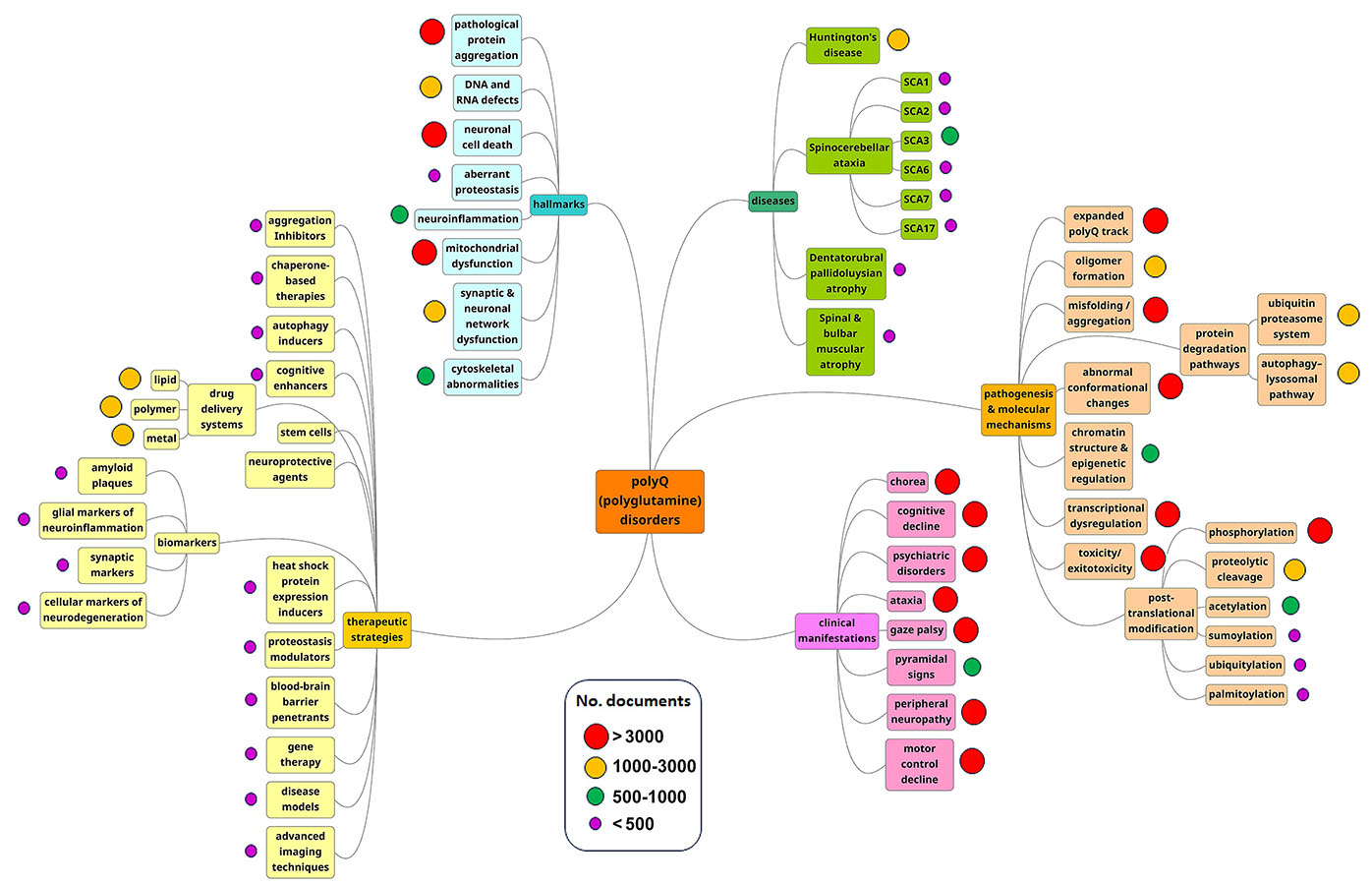
Comme le démontre la carte conceptuelle de notre analyse, la pathogenèse et les mécanismes moléculaires sont les domaines de recherche majeurs (voir figure 3). Ces tendances semblent indiquer que les chercheurs s'efforcent toujours de comprendre le mécanisme du développement de ces maladies chez les patients et dans les familles.
Des stratégies thérapeutiques offrent des percées possibles
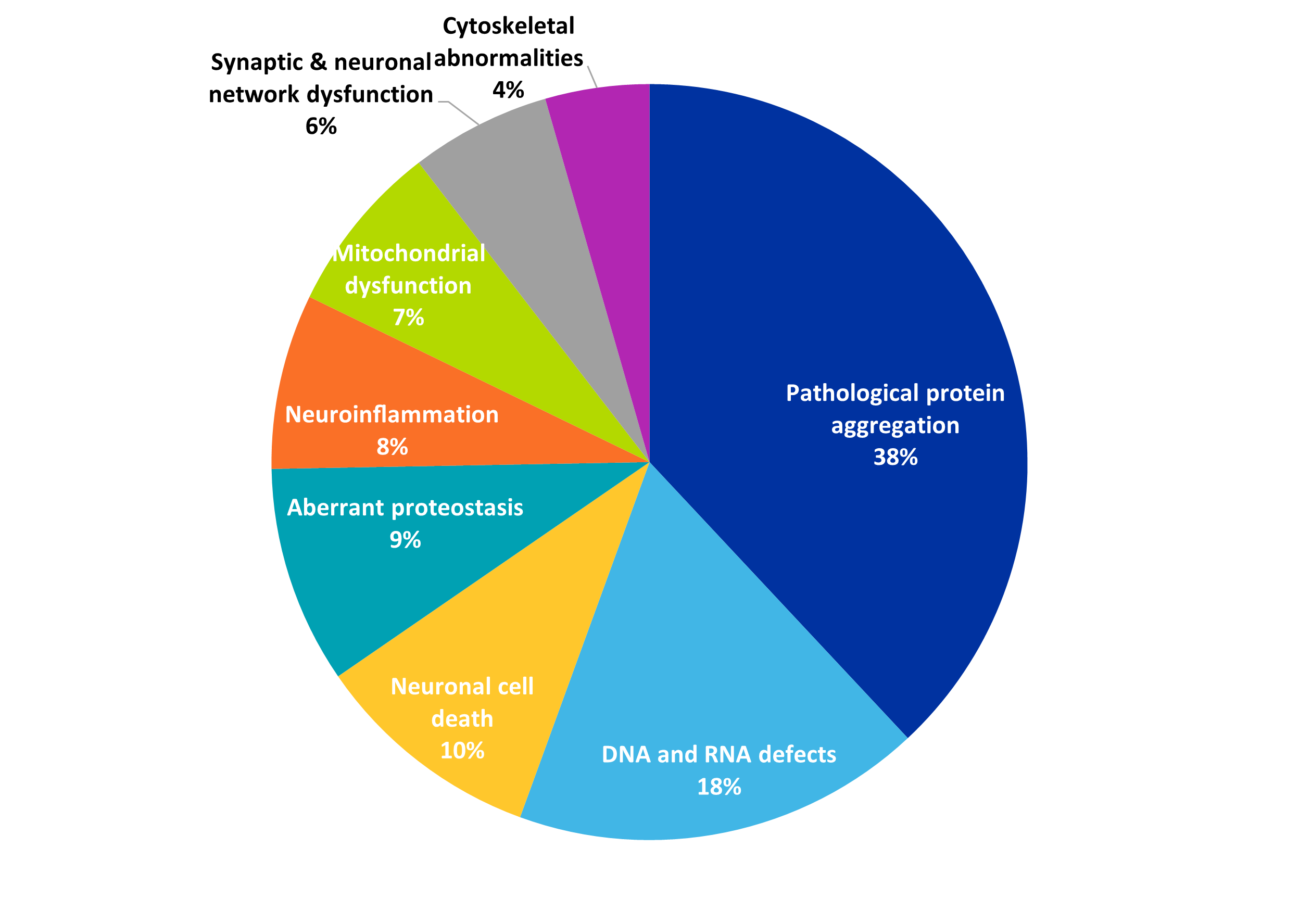
Les traitements et thérapies potentiels sont en développement, tandis que les chercheurs comprennent de mieux en mieux l'agrégation des protéines et les autres attributs des maladies neurodégénératives. Comme on peut le voir sur la figure 4, l'agrégation pathologique des protéines est le signe le plus fréquent de la maladie dans les publications relatives aux maladies par expansion de polyglutamine et, logiquement, les recherches sur ce phénomène portent concomitamment sur les inhibiteurs de l'agrégation en tant que stratégie thérapeutique.
Plusieurs concepts de recherche prometteurs occupent une place importante dans les publications sur les maladies par expansion de polyglutamine :
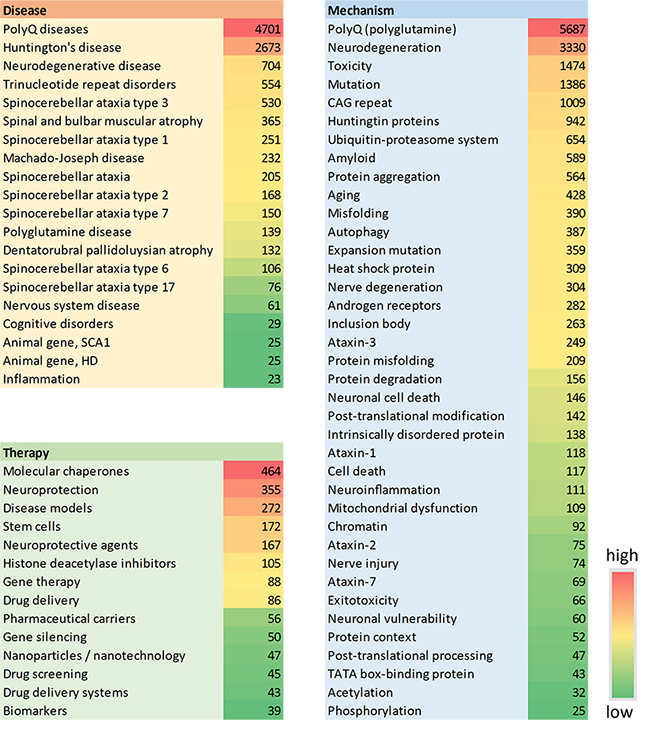
- Chaperons moléculaires : ces éléments jouent un rôle clé dans le maintien de la protéostase cellulaire en facilitant le repliement correct des protéines cellulaires pour garantir leur fonction ou en faisant progresser la dégradation des protéines incurablement mal repliées pour éviter les dommages. Le traitement par des chaperons est une stratégie thérapeutique développée récemment pour traiter les maladies du repliement des protéines, et certains chaperons, notamment ceux de la famille Hsp70, ont démontré leur capacité à modifier l'agrégation des polyglutamines et inhiber leur toxicité. Comprendre comment les chaperons moléculaires interagissent avec la progression de la maladie sera crucial pour de futures avancées, et ce concept est actuellement la thérapie la plus explorée dans les documents de CAS collection de contenus (voir figure 5).
- Thérapie par cellules souches : les cellules souches constituent un outil prometteur pour la médecine régénérative, et une nouvelle approche des maladies par expansion de polyglutamine consiste à remplacer les neurones endommagés ou à stimuler les chemins de la neurogenèse endogène du cerveau en les utilisant. Des tentatives de transplantation de cellules souches ont déjà été réalisées avec succès dans des modèles de maladies par expansion de polyglutamine. Comme on peut le voir sur la figure 5, les cellules souches sont la quatrième thérapie la plus fréquente dans les publications liées aux maladies par expansion de polyglutamine, et elles jouent un rôle majeur dans les modèles de la maladie, qui se classent en troisième position.
- Modèles cellulaires : ces éléments sont essentiels pour la recherche sur la maladie, et notre analyse a noté le développement de modèles de cellules souches embryonnaires et induit des modèles de cellules souches pluripotentes comprenant des informations génétiques spécifiques au patient. Ces avancées génétiques personnalisées pourraient devenir des percées majeures dans le traitement des maladies par expansion de polyglutamine.
- Inhibiteurs de l'histone désacétylase (HDAC) : le statut d'acétylation de la chromatine est totalement perturbé par les troubles par expansion du polyglutamine et l'hyperacétylation des histones déclenchée par l'inhibition des HDAC possède un effet neuroprotecteur. Les inhibiteurs de HDAC à petites molécules présentent donc un potentiel en tant qu'agents thérapeutiques, mais ils s'accompagnent de préoccupations en termes de toxicité. Des études supplémentaires sont nécessaires pour comprendre pleinement le mécanisme de certains inhibiteurs de HDAC bénéfiques, les substrats impliqués et les chemins des enzymes HDAC spécifiques.
- Nanoparticules : alors que les vecteurs traditionnels d'administration des médicaments ont du mal à franchir la barrière hémato-encéphalique, les nanovecteurs peuvent être utiles pour encapsuler les médicaments et cibler leur administration dans ces maladies neurodégénératives difficiles. L'absorption des nanoparticules par les cellules est nettement supérieure à celle des particules plus grosses, et un certain nombre de matériaux produisent des nanovecteurs prometteurs, notamment des polymères, des lipides, des métaux ou des combinaisons de ces matériaux.
Par exemple, les nanoparticules lipidiques solides délivrant un neuroprotecteur ont été administrées avec succès dans des essais sur les rongeurs en lien avec la maladie de Huntington. Dans une autre étude, l'agrégation de Huntington mutant contenant du polyglutamine a également été perturbée dans les cellules neuronales de cerveaux de souris atteintes de la maladie de Huntington en utilisant un système de nanovecteurs enrobés de polyacrylate Fe2O3 et de poly(tréhalase) conjugués de manière covalente.
- Biomarqueurs : une combinaison de biomarqueurs et d'évaluation clinique peut faciliter le diagnostic ainsi que l'évaluation des traitements potentiels. Les chercheurs explorent les biomarqueurs des maladies par expansion de polyglutamine qui ne sont pas rassemblés post-mortem ou par des mesures excessivement invasives, et les avancées dans ce domaine amélioreront considérablement le diagnostic, offriront de meilleures informations de pronostic et faciliteront l'analyse de la progression de la maladie et des traitements. Ces biomarqueurs potentiels comprennent des molécules hépatiques telles que des fragments de protéines mutantes ou des micro-ARN, la neuro-imagerie, comme les IRM, les marqueurs liés au stress oxydatif et à l'inflammation et des modificateurs génétiques via le séquençage du génome.
- Traitements génétiques : les thérapies géniques pourraient un jour inclure le silençage génique ou la suppression du ou des gènes mutés qui provoquent une perturbation du polyglutamine. Ces traitements ont produit des résultats prometteurs lors d'essais sur des animaux pour l'AMB, le SCA1 et la maladie de Huntington, mais les technologies de modification génique suscitent un certain nombre de préoccupations éthiques et logistiques qui devront être traitées avant qu'elles ne soient plus largement adoptées.
Les essais cliniques sont peu nombreux, mais en augmentation
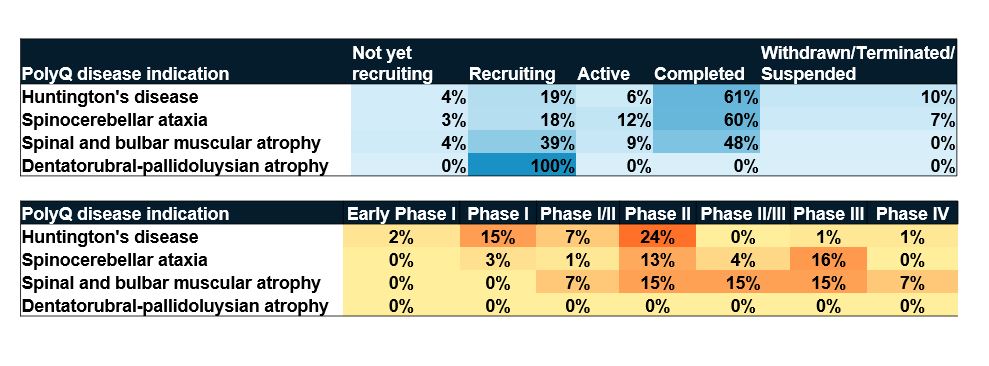
La relative rareté des essais cliniques sur les maladies par expansion de polyglutamine témoigne du fait que ces maladies elles-mêmes sont relativement rares. Notre analyse indique qu'environ 200 essais cliniques ont été enregistrés au cours des 10 dernières années. À titre de comparaison, le National Institute on Aging (Institut national sur le vieillissement) organise actuellement plus de deux fois plus d'études sur la maladie d'Alzheimer. Toutefois, de nombreux essais cliniques sont en phase de recrutement, ce qui signifie que des avancées pourraient se profiler à l'horizon pour ces maladies difficiles à traiter (voir figure 6).
L'avenir de la recherche sur les polyglutamines
La maladie de Huntington et d'autres troubles liés aux polyglutamines sont des affections complexes présentant de nombreux obstacles au développement de stratégies thérapeutiques efficaces. Toutefois, à mesure que notre compréhension des procédés neurodégénératifs s'améliore, les chances de réaliser des progrès cliniques majeurs dans le traitement de ces maladies progressent également.
Notre analyse de CAS Collection de contenus démontre que les orientations futures de la recherche porteront sur différents domaines potentiels. Le développement de nouvelles stratégies pour prévenir ou séparer les agrégats de protéines toxiques sera essentiel. La compréhension des modificateurs génétiques et la définition d'approches de thérapie génique pourraient libérer de nombreux traitements ou remèdes et l'identification des biomarqueurs pour un diagnostic et une évaluation de traitement plus clairs fournira aux médecins davantage d'options pour gérer la maladie.
Compte tenu de la complexité des maladies par expansion de polyglutamine, il sera important pour les chercheurs d'explorer les effets synergiques potentiels de la combinaison de différentes approches thérapeutiques. Un succès permettrait de ralentir, voire d'arrêter certaines des maladies neurodégénératives les plus complexes et les plus affaiblissantes qui affectent la société actuelle.