Gain new perspectives for faster progress directly to your inbox.
Neurodegenerative diseases critically harm the lives of millions worldwide. These complex disorders involve numerous genetic and non-genetic factors, but they all feature pathological protein aggregation or issues with proteostasis, the process of protein synthesis, folding, disaggregation, and degradation. The most well-known neurodegenerative diseases are Alzheimer’s and Parkinson’s, which have become more common as people live longer and consequently undergo the effects of aging. However, there is another group of neurodegenerative disorders known as polyglutamine (PolyQ) diseases, including Huntington’s disease, that present additional challenges to researchers pursuing treatment options.
PolyQ diseases are characterized by the abnormal expansion of a cytosine-adenine-guanine (CAG) trinucleotide repeat, resulting in the production of proteins with an expanded polyglutamine tract. They are considered rare, with an average frequency between 1-10 cases per 100,000 people, but PolyQs are the largest group of monogenic neurodegenerative disorders. With neither a cure nor preventive measures available, these diseases are complicated to treat and can severely impact entire families because of their genetic nature.
We analyzed the CAS Content CollectionTM, the world’s largest human-curated repository of scientific information, to better understand current research breakthroughs and ongoing challenges to addressing these life-threatening diseases.
What are PolyQ diseases?
There are nine identified PolyQ diseases: Huntington's disease (HD); six spinocerebellar ataxias (SCA) types one, two, three, six, seven, and seventeen; dentatorubral pallidoluysian atrophy (DRPLA); and spinal and bulbar muscular atrophy (SBMA). All of these conditions are autosomal dominant except SBMA, which is X-linked and only occurs in male populations.
PolyQ diseases exhibit an inverse correlation between the number of CAG repeats and the age of patients at the onset of disease. The disease’s severity tends to increase, and the onset age tends to decrease in successive generations.
The proteins involved in the various disorders differ in their function and location within cells, and different brain regions and neuronal subtypes are affected in each condition. However, a common feature of these diseases is the radical deformation of neurons in specific regions of the brain, which impairs vital functioning.
Challenges in treating Huntington’s disease and other PolyQ conditions
Currently, there are no disease-modifying treatments available for Huntington’s disease or other PolyQs, and several ongoing challenges have complicated the search for cures:
- Complex pathophysiology: PolyQ diseases involve intricate molecular mechanisms with many complexities that researchers are working to more fully understand.
- Variable clinical presentation: These conditions exhibit significant heterogeneity in presentation, age of onset, and disease progression. This complicates diagnosis and treatment.
- Lack of biomarkers for diagnosis and monitoring: Reliable biomarkers are critical for diagnosis, prognosis, and monitoring treatment responses, but validated biomarkers for PolyQ diseases are limited.
- Multiple drug development hurdles: Animal models do not fully represent the human disease phenotype of these conditions, so their value is limited in PolyQ research. At the same time, potential treatments struggle to penetrate the blood-brain barrier and reach target neurons, all of which complicates the development of effective therapies.
Research is increasing, but much work remains
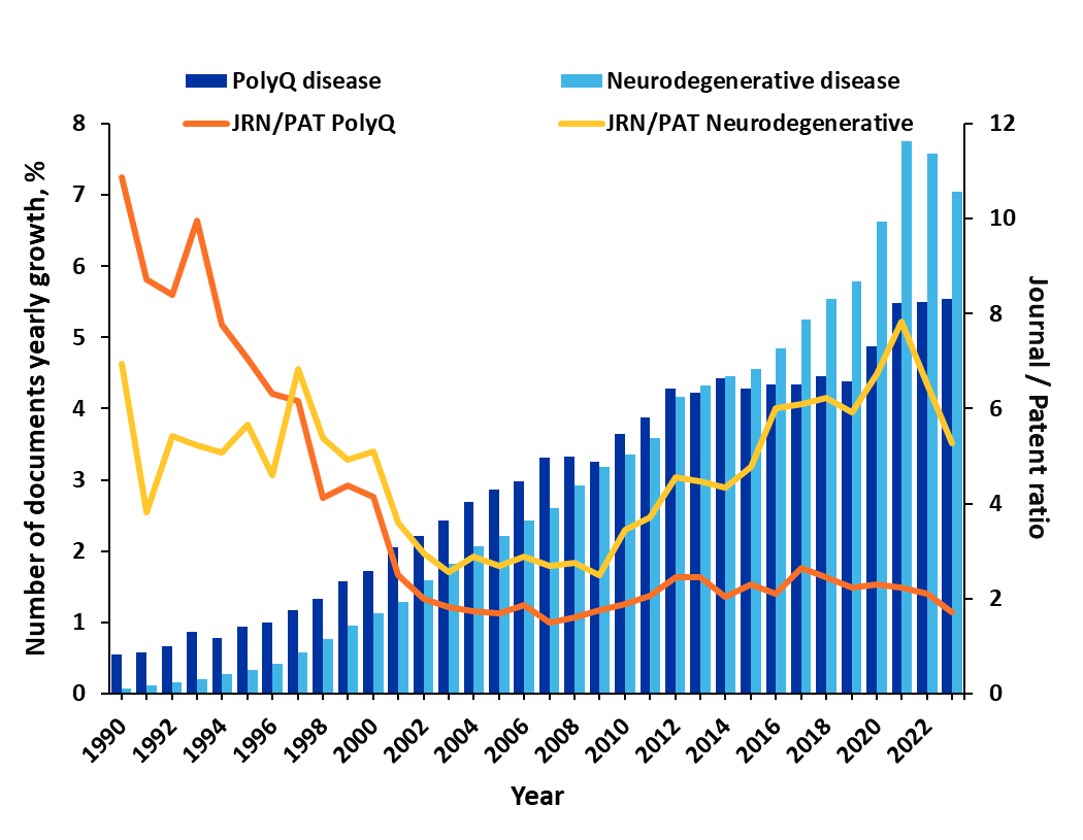
Despite these many challenges, researchers are pursuing a better understanding of these diseases and potential treatments. Our analysis of the CAS Content Collection revealed there has been more than 25% growth in PolyQ-related publications in just the last three years. Notably, in earlier years, journal publications dominated the research with a journal-to-patent ratio of 4-5, but after the year 2002, patent publications grew significantly, and the ratio is now closer to 2 (see Figure 1). This suggests that therapeutic discoveries are moving closer to commercialization.
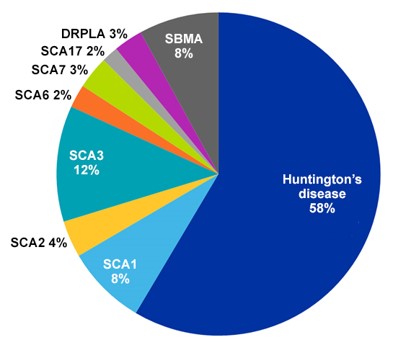
More than half of all PolyQ-related documents in the CAS Content Collection pertain to Huntington’s disease, which makes sense since this is the most prevalent PolyQ condition with an average frequency of three to seven cases per 100,000 people (see Figure 2). Chorea is the most common symptom of Huntington’s disease, but it also leads to cognitive changes and dementia. It’s triggered by a CAG repeat expansion of the exon of the huntington gene.
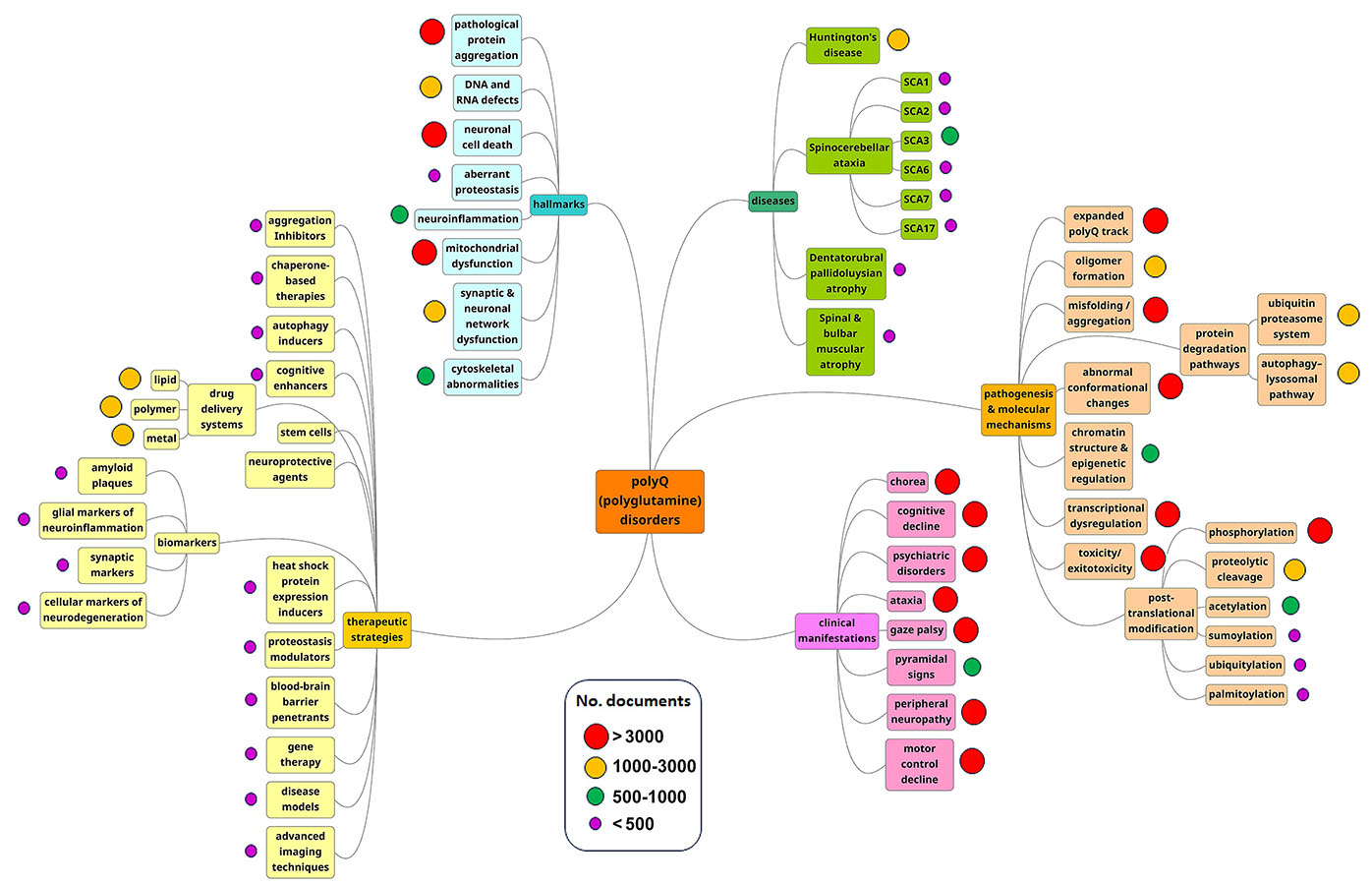
As the mind map of our analysis demonstrates, pathogenesis and molecular mechanisms are the most prominent research areas (see Figure 3). These trends suggest that researchers are still seeking to understand how these diseases develop in patients and families.
Therapeutic strategies offer possible breakthroughs
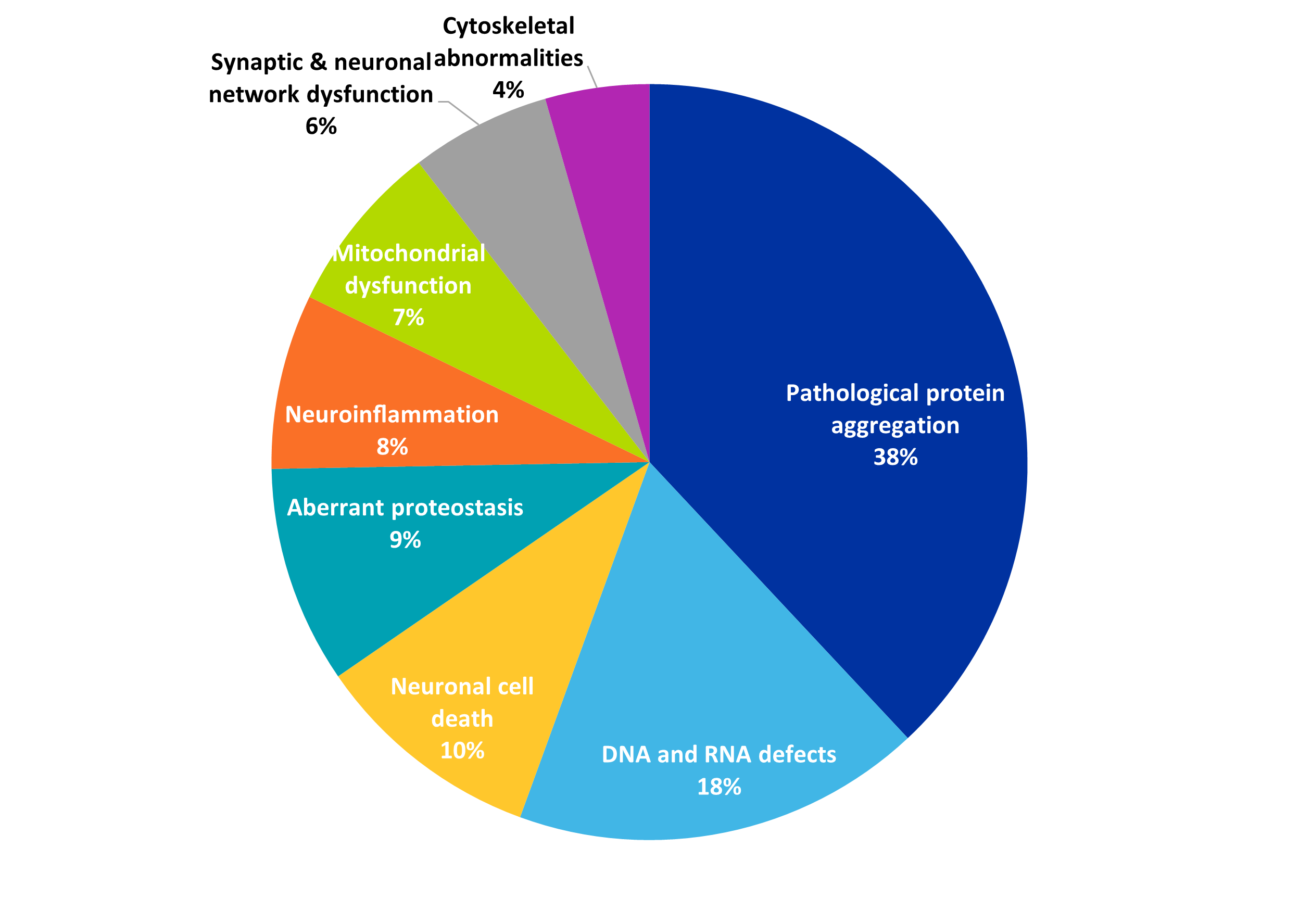
Potential treatments and therapies are growing, as researchers gain a better understanding of protein aggregation and other attributes of neurodegenerative diseases. As we see in Figure 4, pathological protein aggregation is the most common disease hallmark in PolyQ-related publications, and it understandably co-occurs with aggregation inhibitors as a therapeutic strategy.
Several promising research concepts are prominent in PolyQ publications are:
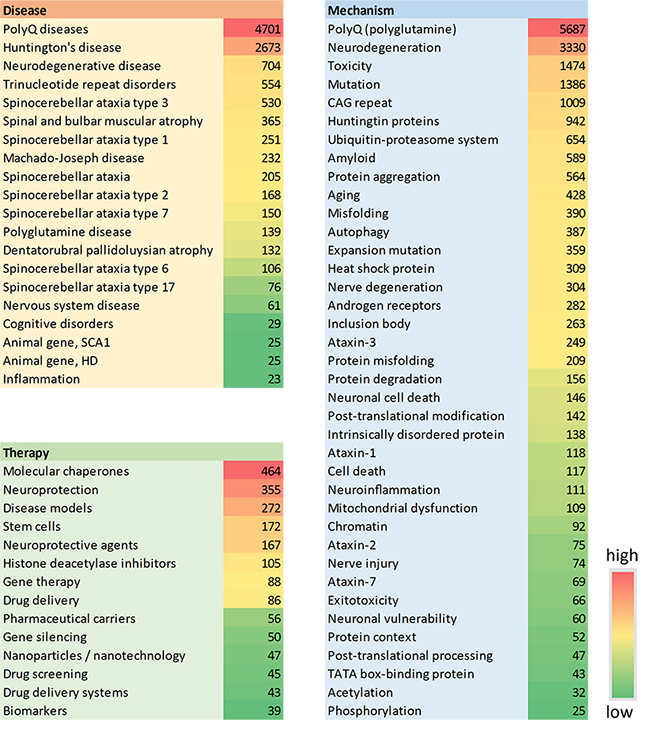
- Molecular chaperones: These play a key role in maintaining cellular proteostasis by facilitating the correct folding of cellular proteins to guarantee their function or by advancing the degradation of terminally misfolded proteins to prevent damage. Chaperone therapy is a recently developed therapeutic strategy to treat protein misfolding diseases, and certain chaperones, such as the Hsp70 family, have been shown to modify PolyQ aggregation and inhibit its toxicity. Understanding how molecular chaperones interrelate with disease progression will be crucial to future breakthroughs, and this concept is currently the most-explored therapy in CAS Content Collection materials (see Figure 5).
- Stem cell therapy: Stem cells are a promising tool for regenerative medicine, and one novel approach to PolyQ diseases is to replace damaged neurons or stimulate the endogenous neurogenesis pathways of the brain using them. There have already been successful stem-cell transplantation attempts in PolyQ models. As seen in Figure 5, stem cells are the fourth most common therapy in PolyQ-related publications, and they also play an important role in disease models, which rank third.
- Cellular models: These are crucial to disease research, and our analysis noted the development of embryonic stem cell models and induced pluripotent stem cell models comprising patient-specific genetic information. These personalized genetic advances could become major breakthroughs in the treatment of PolyQ diseases.
- Histone deacetylase (HDAC) enzyme inhibitors: Chromatin acetylation status is critically impaired in PolyQ disorders, and histone hyperacetylation triggered by the inhibition of HDACs has a neuroprotective effect. Small molecule HDAC inhibitors are therefore showing potential as therapeutic agents, but there are toxicity concerns. Additional studies are needed to fully understand the mechanism of certain beneficial HDAC inhibitors, the substrates involved, and the pathways of specific HDAC enzymes.
- Nanoparticles: While traditional drug delivery vectors struggle to pass through the blood-brain barrier, nanocarriers may be useful for encapsulating drugs and targeting delivery in these challenging neurodegenerative diseases. The uptake of nanoparticles by cells is much higher than larger particles, and a variety of materials make promising nanocarriers, including polymers, lipids, metals, or combinations of these materials.
For example, solid lipid nanoparticles delivering a neuroprotector were successfully administered in rodent trials relating to Huntington’s disease. In another study, aggregation of PolyQ-containing mutant huntingtin was also hindered in neuronal cells in Huntington's disease mouse brains by using an Fe2O3 polyacrylate-coated and covalently conjugated poly(trehalose) nanocarrier system.
- Biomarkers: A combination of biomarkers and clinical assessment can facilitate diagnosis as well as the evaluation of potential therapies. Researchers are exploring biomarkers for PolyQ diseases that are not gathered post-mortem or through overly invasive measures, and breakthroughs in this concept will greatly improve diagnosis, provide better prognosis information, and facilitate the analysis of disease progression and treatments. These potential biomarkers include blood-based molecules such as mutant protein fragments or microRNAs, neuroimaging like MRI scans, markers related to oxidative stress and inflammation, and genetic modifiers via genome sequencing.
- Genetic therapies: Genetic therapies may one day include gene silencing or suppressing the mutated gene(s) that causes a PolyQ disorder. These therapies have produced promising results in animal trials for SBMA, SCA1, and Huntington’s disease, but there are a number of ethical and logistical concerns with gene-editing technologies that must be addressed before there is wider adoption.
Clinical trials are few but growing
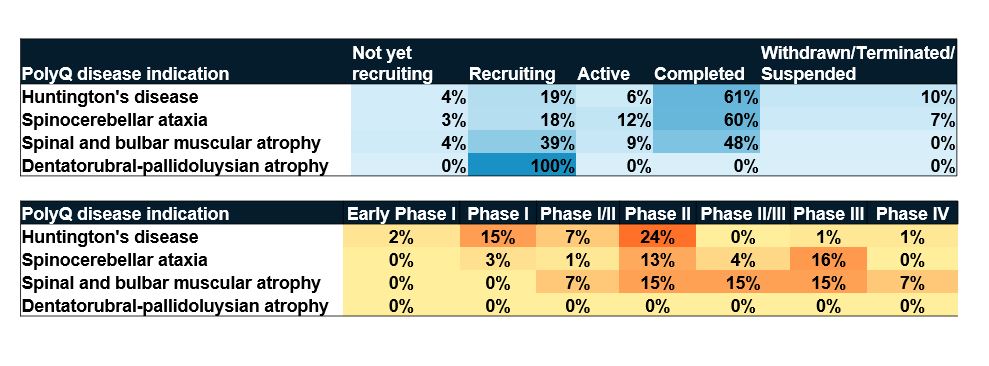
The relative scarcity of clinical trials for PolyQ diseases reflects their relative rarity. Our analysis shows that about 200 clinical trials have been registered over the last 10 years. For comparison, the National Institute on Aging currently has more than double that number for Alzheimer’s today. However, numerous clinical trials are in recruitment status, which means breakthroughs may be on the horizon for these hard-to-treat diseases (see Figure 6).
The future of PolyQs research
Huntington’s disease and other PolyQ conditions are complex disorders with many challenges to the development of successful therapeutic strategies. However, as our understanding of neurodegenerative processes improves, so too will the chances of significant clinical advances in treating these diseases.
Our analysis of the CAS Content Collection shows that future research directions have several potential areas of emphasis. Developing novel strategies to prevent or separate toxic protein aggregates will be critical. Understanding genetic modifiers and defining gene therapy approaches could unlock many treatments or cures, and identifying biomarkers for clearer diagnosis and treatment evaluation will provide clinicians with more disease management options.
Given the complexity of PolyQ diseases, it will be important for researchers to explore the potential synergistic effects of combining different therapeutic approaches. Success could mean slowing or stopping some of the most challenging, debilitating neurodegenerative diseases impacting society today.
Key data in this article, as well as Figures 1-6, are extracted from Tenchov R, Sasso JM, Zhou QA. Polyglutamine (PolyQ) Diseases: Navigating the Landscape of Neurodegeneration. ChemRxiv. 2024; doi:10.26434/chemrxiv-2024-pt7wp.