Gain new perspectives for faster progress directly to your inbox.

Since the World Health Organization declared COVID-19 a pandemic, researchers have learned a tremendous amount about SARS-CoV-2, the new coronavirus that causes this disease. However, despite extensive effort and investment, effective therapeutic treatments for COVID-19 patients have been elusive. Though multiple vaccine candidates have already entered clinical trials globally, even if they prove safe and effective, many months or even years will be required to manufacture and distribute the vaccine and inoculate the global population. Thus, there remains an urgent need to identify effective antiviral treatments that can mitigate the virus’ impact on the many more who will become ill before the pandemic is brought under control.
Scientists have been exploring various ways to accelerate the drug development process to meet this urgent need, including using computational approaches to identify drugs already approved for other indications that may be effective in treating COVID-19. To aid that effort, a group of scientists and technologists at CAS sought to identify possible drug candidates for treating COVID-19 with machine learning models for priority protein targets of SARS-CoV-2 using a Quantitative Structure-Activity Relationship (QSAR) methodology. This work, which successfully identified a number of drugs now beginning to show clinical efficacy, including Lopinavir and Telmisartan, was recently published in ACS Omega.
Something old, something new
Given the substantial time and cost needed to bring a new drug to market, repurposing existing small-molecule drugs is an attractive alternative, especially when the need is so urgent. In addition to getting treatments to market faster, this strategy offers a number of advantages over the traditional drug development process, including lowering the risk of late-stage failure due to negative side effects.
Drug repurposing is not a new concept. However, its application to date has been mostly opportunistic rather than systematic. In some of the most successful examples of drug repurposing so far, such as Viagra and Minoxidil, new indications arose when patients reported unexpected side effects. Recently, more systematic approaches to drug repurposing have been introduced including computational methods such as signature matching, molecular docking, genetic association, pathway mapping and retrospective clinical analysis. It is hoped that a computational approach will allow researchers to reliably connect existing small-molecule therapeutics to newly identified drug targets, maximizing the therapeutic value of existing portfolios.
Closing in on a target
Coronaviruses are a large family of viruses long known to cause mild to moderate upper-respiratory illnesses in humans and many different animal species. Though it is rare for animal-specific coronaviruses to infect and spread in humans, to date three coronaviruses have proven able to make that jump: SARS-CoV-1, MERS-CoV and the new SARS-CoV-2. All three are beta-coronaviruses believed to have originated in bats. Given the similarities between these viruses and their progress to human contagion, previous SARS and MERS research provides a good starting point when seeking drugable targets for SARS-CoV-2. Among all the proteins in SARS-CoV-2, the 3-chymotrypsin-like protease (3CLpro) and RNA-dependent RNA polymerase (RdRp) are two ideal protein targets for QSAR modeling, in part due to significant similarities they share with proteins identified in SARS-CoV and MERS-CoV as well as other known coronaviruses.
3CLpro is a protease that is required for the coronavirus to cleave the polyprotein peptides into individual functional non-structural proteins (NSPs). When comparing amino acid sequences and protein structures, 3CLpro was found to be highly conserved between SARS-CoV-2 and other human coronaviruses. It shows a 96% sequence identity overlap with SARS-CoV-1, 87% with MERS-CoV, and 90% with Human-CoV. Therefore, the 3CLpro inhibitors identified in previous coronavirus-related research are promising inhibitors for SARS-CoV-2 3CLpro, and the associated structure-activity relationship (SAR) data are valuable for training machine learning models searching for new inhibitors of SARS-CoV-2 3CLpro.
RdRp is the major enzyme utilized by RNA viruses to replicate viral genomes in host cells. Structural study and sequence analysis of SARS-CoV-2 RdRp revealed that this enzyme is very similar to the structure of SARS-CoV-1 RdRp and contains several key amino acid residues that are conserved in most viral RdRps, including HCV. Fortunately, various viral RdRps have been widely studied as inhibitors of RNA viruses, especially in HCV-related research. Therefore, existing RdRp inhibitors for the RNA viruses, such as HCV, may provide valuable insights for drug development for SARS-CoV-2 RdRp inhibition.
Prioritizing existing therapeutics with machine learning
Machine learning models have increasingly been used to facilitate drug discovery in recent years. Specifically, QSAR is often one of the first steps in the modern drug discovery process. Simply put, QSARs are mathematical models approximating rather complicated biological or physicochemical properties of chemicals based on quantitative measures of their molecular structures. These predictive mathematical models are used for screening large databases of chemical structures to prioritize potential drug candidates that are most likely to be active against identified targets. This approach assumes that the activity of a chemical substance is directly related to its structure, and thus, molecules with similar structural features will exhibit similar physical properties and/or biological effects.
In this study, my colleagues and I closely collaborated to build highly predictive QSAR models for 3CLpro and RdRp protein targets. The team, which included computational scientists and chemists, curated more than 1,000 inhibitors with structure-bioactivity data as training molecules for the models. We collected data from the most current SARS-CoV-2 bioassay studies, as well as existing studies with SARS-CoV-1, MERS-CoV and other related viruses in the CAS content collection. Using these data, we applied a variety of machine learning algorithms to build several dozen QSAR models – selecting from among these the strongest performing models – one targeting 3CLpro and one targeting RdRp.
Read the full journal article QSAR machine learning models and their applications for identifying viral 3CLpro- and RdRp-targeting compounds as potential therapeutics for COVID-19 and related viral infections to see all the models tested and which potential candidates rose to the top.
We used the two resulting QSAR models to screen a large pool of potential drug candidates including 1,087 FDA-approved drugs, nearly 50,000 substances from the CAS COVID-19 Antiviral Candidate Compounds Dataset and ~113,000 substances with pharmacological activity identified or a therapeutic role indexed by CAS in SARS-, MERS- and COVID-19-related documents published since 2003. By modeling protease inhibitor activity as a function of substance structure, we identified some of the most promising candidates among substances predicted to be active inhibitors of coronavirus 3CLpro and RdRp. Additionally, a number of the substances that our models predict will inhibit 3CLpro or RdRp in SARS-CoV-2 also have previously identified therapeutic activity against other diseases that have emerged as risk factors for more severe COVID-19 infections. For example, a candidate COVID-19 antiviral that also has known activity against heart disease, such as diltiazem hydrochloride (Cardizem), could potentially provide a dual benefit, in certain cases.
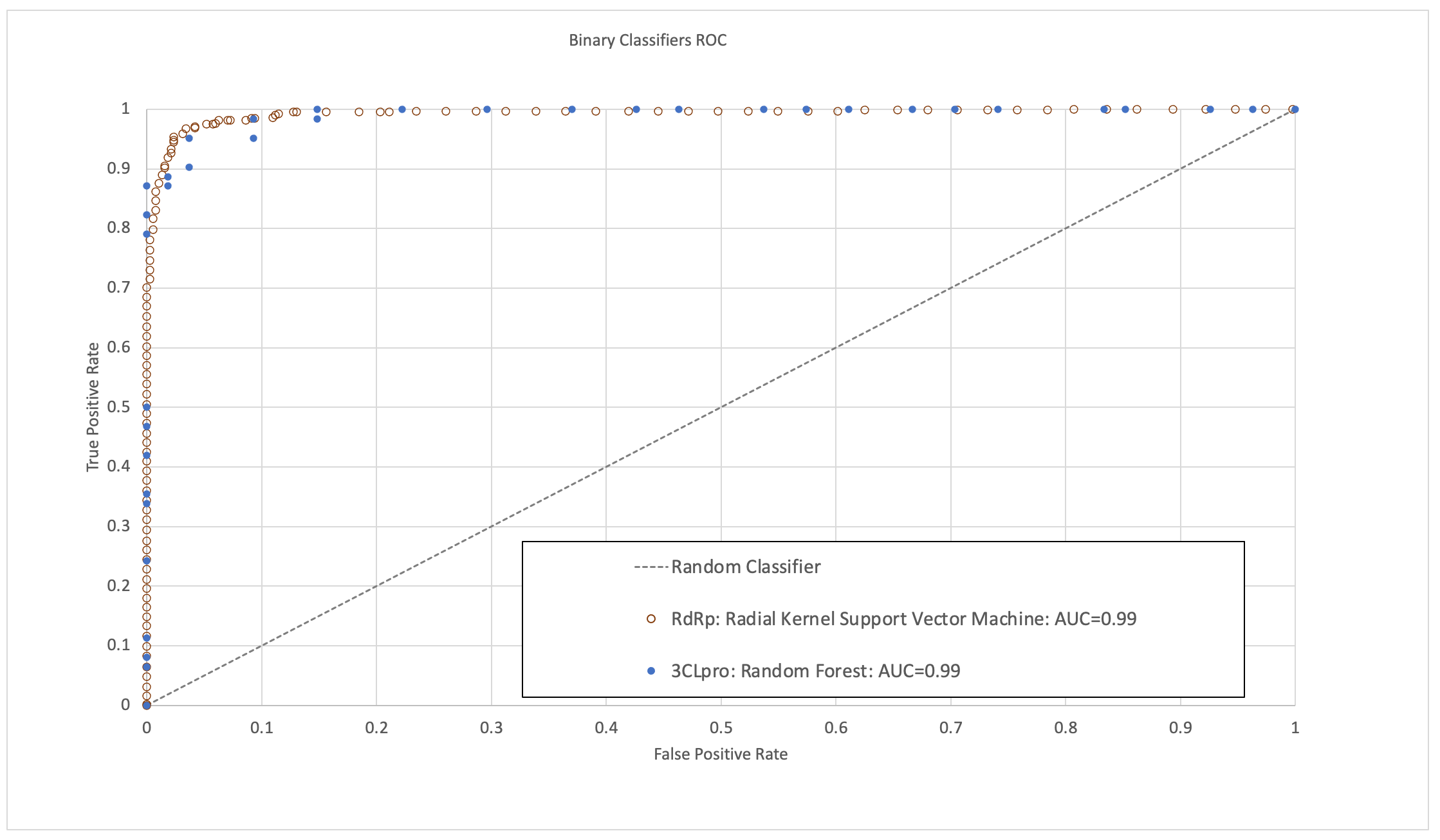
The models were validated to have high area under the receiver operating characteristic curve (ROC-AUC), sensitivity, specificity and accuracy (Figure 1). In the time since this research was completed, some molecules predicted to have high activity by these models have now been validated by published experimental bioassay studies and clinical trials, providing further positive indication of their predictive ability.
Getting ahead of the next pandemic
While this study was focused on identifying potential therapeutic compounds for use in the current COVID-19 crisis, it is likely there will be additional pandemics of viral origin in the years to come. Thus, it is urgent that preparation for future outbreaks begins now with continued investment and focus on antiviral agent research. Because different types of viruses can cause epidemics (e.g., coronavirus, influenza viruses, Ebola viruses, retroviruses) and human safety and efficacy testing for each new drug or indication still takes significant time, broad-spectrum antiviral agents and vaccines would be of greatest value.
The ongoing development of computer-based drug discovery methods, such as the machine learning procedures described here, molecular docking and virtual screening, will be of central importance. The ongoing increase in computer processing power and continued development of docking and structure prediction algorithms and protein crystal structure determination techniques will facilitate progress. Additionally, the use of high-throughput screening, omics technologies and the repurposing of already-developed drugs will continue and increase in importance. However, these new technology-driven methods won’t replace human laboratory research, but will instead complement it through increased efficiency. We hope this effort, which combined human data curation and machine learning models to successfully identify potential small-molecule drug candidates for COVID-19, highlights the value of synergy between humans and machines in drug discovery, while contributing to on-going antiviral research efforts for COVID-19 and beyond.
As part of the global scientific community, CAS is committed to leveraging all of our assets and capabilities to support the fight against COVID-19. Explore our additional open-access CAS COVID-19 resources including scientific insights, open access datasets and special reports.